Written 11/13/2006 by Liberty Hamilton. Updated 2/17/08 by Owen Phillips. Email Dr. Katherine Narr if you have any questions.
To run R, you will need the ucf files for each subject as well as a text file describing each subject. The ucf files and the text file should be placed in the same directory.
To run on the command line, you must first make sure that your account is set up to use R on cerebro (the grid). To do this, follow these steps (you should only have to do this once!):
File group sex age genotype brain_vol
The first column should contain the full names of the ucfs that you are analyzing. This can include the path, but if you run R in the same directory as your UCFs you will not need the path name to be specified.
Remember: You will need separate text files if you want to look at male controls versus male patients, or female controls versus female patients. All of the files in the text file will be used in the analysis; there isn't a way to specify only certain lines in the text file.
Example Text File: (note that columns don't line up, but there is a tab between each field)
File group sex age genotype brain_vol
/mydirectory/project/1234_hippo_L.ucf 1 0 23 aa 1803
/mydirectory/project/1254_hippo_L.ucf 0 1 42 bb 1799
/mydirectory/project/1412_hippo_L.ucf 1 1 55 ab 1782
/mydirectory/project/1223_hippo_L.ucf 1 0 29 bb 1833
/mydirectory/project/1255_hippo_L.ucf 0 1 42 aa 1792
/mydirectory/project/4921_hippo_L.ucf 0 0 24 ab 1822
Running R on the command line is pretty simple, even though the command line arguments are very long. To run on the grid, be sure to put the "qsub" command before your command line so that your job will be submitted to the cluster queue.
Here is an example:
qsub -b y -q long.q /ifs/woods/R/R-2.7.2/bin/R CMD BATCH --no-save --no-restore --quiet --args -table/mydirectory/project/text_file_LEFT.txt -formulay~sex+age+group+genotype -reducedy~sex+age+genotype -output/mydirectory/project/L_groupeffect.ucf /ifs/woods/rshape/batch_commands/anova_shape_with_sign_batch.R /mydirectory/project/L_groupeffect_error.log
So, you will have to change these parts: -table, -formulay, -reducedy, -output
| -table | The text file you created in the first section. |
| -formulay | All of the variables you want to look at (all headers in your text file, excluding File) |
| -reducedy | Your covariates, i.e. the variables for which you don't want to look at effects. If you want to see group effects, put everything but group in your -reducedy. |
| -output | The name of the ucf file to be outputted. Call this something like group_effects_L.ucf or groupfemalesL.ucf. |
Also remember to change the error log at the very end, so you will have an error log for each instance of R that you run. You can use this error log to see where things may have gone wrong.
qsub -b y -q long.q /ifs/woods/R/R-2.7.2/bin/R CMD BATCH --no-save --no-restore --quiet --args -table/mydirectory/project/text_file_LEFT.txt -formulay~sex+age+group+genotype -reducedy~age+group+genotype -output/mydirectory/project/L_sexeffect.ucf /ifs/woods/rshape/batch_commands/anova_shape_with_sign_batch.R /mydirectory/project/L_sexeffect_error.log
qsub -b y -q long.q /ifs/woods/R/R-2.7.2/bin/R CMD BATCH --no-save --no-restore --quiet --args -table/mydirectory/project/text_file_LEFT.txt -formulay~sex+age+group+genotype+group:genotype -reducedy~sex+age+group+genotype -output/mydirectory/project/L_groupbygenoeffect.ucf /ifs/woods/rshape/batch_commands/anova_shape_with_sign_batch.R /mydirectory/project/L_groupbygenoeffect_error.log
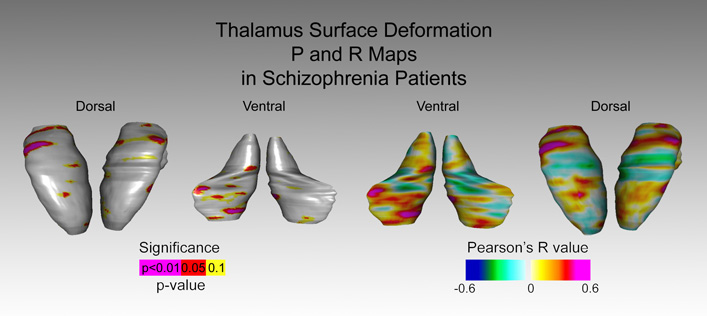
To run an ANOVA in R using the Pipeline, download the anova_signed.pipe module and create the text files as detailed above.
The following statistical models are also possible using the scripts found in /ifs/woods/rshape/batch_commands.